Variance partitioning the bulk RNAseq data
Anthony Hung
2021-01-19
Last updated: 2021-01-21
Checks: 7 0
Knit directory: invitroOA_pilot_repository/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20210119) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 58e93fd. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: code/bulkRNA_preprocessing/.snakemake/conda-archive/
Ignored: code/bulkRNA_preprocessing/.snakemake/conda/
Ignored: code/bulkRNA_preprocessing/.snakemake/locks/
Ignored: code/bulkRNA_preprocessing/.snakemake/shadow/
Ignored: code/bulkRNA_preprocessing/.snakemake/singularity/
Ignored: code/bulkRNA_preprocessing/.snakemake/tmp.3ekfs3n5/
Ignored: code/bulkRNA_preprocessing/fastq/
Ignored: code/bulkRNA_preprocessing/out/
Ignored: code/single_cell_preprocessing/.snakemake/conda-archive/
Ignored: code/single_cell_preprocessing/.snakemake/conda/
Ignored: code/single_cell_preprocessing/.snakemake/locks/
Ignored: code/single_cell_preprocessing/.snakemake/shadow/
Ignored: code/single_cell_preprocessing/.snakemake/singularity/
Ignored: code/single_cell_preprocessing/YG-AH-2S-ANT-1_S1_L008/
Ignored: code/single_cell_preprocessing/YG-AH-2S-ANT-2_S2_L008/
Ignored: code/single_cell_preprocessing/demuxlet/.DS_Store
Ignored: code/single_cell_preprocessing/fastq/
Ignored: data/external_scRNA/Chou_et_al2020/
Ignored: data/external_scRNA/Jietal2018/
Ignored: data/external_scRNA/Wuetal2021/
Ignored: data/external_scRNA/merged_external_scRNA.rds
Ignored: data/poweranalysis/alasoo_et_al/
Ignored: output/GO_terms_enriched.csv
Ignored: output/topicModel_k=7.rds
Ignored: output/voom_results.rds
Unstaged changes:
Modified: .gitignore
Modified: data/ANT1_2.rds
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/variation_bulkRNA.Rmd) and HTML (docs/variation_bulkRNA.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | e3fbd0f | Anthony Hung | 2021-01-21 | add ward et al data files to data |
| html | e3fbd0f | Anthony Hung | 2021-01-21 | add ward et al data files to data |
| Rmd | 0711682 | Anthony Hung | 2021-01-21 | start normalization file |
| Rmd | 28f57fa | Anthony Hung | 2021-01-19 | Add files for analysis |
Introduction
This code uses the variancePartition package to quantify the percent of variance in the data explained by some biological/technical factors.
Load packages and normalized data
library(variancePartition)Loading required package: ggplot2Loading required package: limmaLoading required package: foreachLoading required package: scalesLoading required package: BiobaseLoading required package: BiocGenericsLoading required package: parallel
Attaching package: 'BiocGenerics'The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLBThe following object is masked from 'package:limma':
plotMAThe following objects are masked from 'package:stats':
IQR, mad, sd, var, xtabsThe following objects are masked from 'package:base':
anyDuplicated, append, as.data.frame, basename, cbind,
colnames, dirname, do.call, duplicated, eval, evalq, Filter,
Find, get, grep, grepl, intersect, is.unsorted, lapply, Map,
mapply, match, mget, order, paste, pmax, pmax.int, pmin,
pmin.int, Position, rank, rbind, Reduce, rownames, sapply,
setdiff, sort, table, tapply, union, unique, unsplit, which,
which.max, which.minWelcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
Attaching package: 'variancePartition'The following object is masked from 'package:limma':
classifyTestsFlibrary(RUVSeq)Loading required package: EDASeqLoading required package: ShortReadLoading required package: BiocParallelLoading required package: BiostringsLoading required package: S4VectorsLoading required package: stats4
Attaching package: 'S4Vectors'The following object is masked from 'package:base':
expand.gridLoading required package: IRangesLoading required package: XVector
Attaching package: 'Biostrings'The following object is masked from 'package:base':
strsplitLoading required package: RsamtoolsLoading required package: GenomeInfoDbLoading required package: GenomicRangesLoading required package: GenomicAlignmentsLoading required package: SummarizedExperimentLoading required package: DelayedArrayLoading required package: matrixStats
Attaching package: 'matrixStats'The following objects are masked from 'package:Biobase':
anyMissing, rowMedians
Attaching package: 'DelayedArray'The following objects are masked from 'package:matrixStats':
colMaxs, colMins, colRanges, rowMaxs, rowMins, rowRangesThe following object is masked from 'package:Biostrings':
typeThe following objects are masked from 'package:base':
aperm, apply, rowsumLoading required package: edgeRlibrary(edgeR)
library(dplyr)
Attaching package: 'dplyr'The following object is masked from 'package:ShortRead':
idThe following objects are masked from 'package:GenomicAlignments':
first, lastThe following object is masked from 'package:matrixStats':
countThe following objects are masked from 'package:GenomicRanges':
intersect, setdiff, unionThe following object is masked from 'package:GenomeInfoDb':
intersectThe following objects are masked from 'package:Biostrings':
collapse, intersect, setdiff, setequal, unionThe following object is masked from 'package:XVector':
sliceThe following objects are masked from 'package:IRanges':
collapse, desc, intersect, setdiff, slice, unionThe following objects are masked from 'package:S4Vectors':
first, intersect, rename, setdiff, setequal, unionThe following object is masked from 'package:Biobase':
combineThe following objects are masked from 'package:BiocGenerics':
combine, intersect, setdiff, unionThe following objects are masked from 'package:stats':
filter, lagThe following objects are masked from 'package:base':
intersect, setdiff, setequal, union#raw(filtered) counts
counts_upperquartile <- readRDS("data/filtered_counts.rds")$counts
#normalized and filtered counts
filtered_upperquartile <- readRDS("data/norm_filtered_counts.rds")
#metadata
sampleinfo <- readRDS("data/Sample.info.RNAseq.reordered.rds")Fitting unwanted variation
Before fitting the linear mixed model to quantify contributions of different variables to variation in the dataset, we are also interested in fitting one factor of unwanted variation to include in the model using RUVg. For the RUVg control genes, we use the top 100 (~1% of the genes considered) LEAST variable genes as control genes.
#input data consists of raw filtered data (filtered for lowly expressed genes)
#compute CV (stdev/mean) and rank from least to most; pick 100 least variable
cv <- apply(counts_upperquartile, 1, function(x) sd(x)/mean(x))
least_var_genes <- names(head(sort(cv), 100))Apply RUVseq with the set of least variable genes to determine a single factor of unwanted variation.
#load data into expressionset
set <- newSeqExpressionSet(as.matrix(counts_upperquartile),phenoData = data.frame(sampleinfo, row.names=colnames(counts_upperquartile)))
setSeqExpressionSet (storageMode: lockedEnvironment)
assayData: 10486 features, 17 samples
element names: counts, normalizedCounts, offset
protocolData: none
phenoData
sampleNames: 18855_3_S 19160_3_S ... 18855_3_U (17 total)
varLabels: Sample_ID Individual ... LibSize (8 total)
varMetadata: labelDescription
featureData: none
experimentData: use 'experimentData(object)'
Annotation: #normalization
set <- betweenLaneNormalization(x = set, which = "upper")
#run RUVg
set1 <- RUVg(set, least_var_genes, k=1)
sample_info <- pData(set1)
sample_info$Replicate <- as.factor(sample_info$Replicate)
sample_info$LibraryPrepBatch <- as.factor(sample_info$LibraryPrepBatch)Specify variables to consider in LMM
Here, we specify two separate models (one including the single factor of unwanted variation computed above and one without this factor).
# Specify variables to consider
form <- ~ (1|Individual) + (1|treatment) + (1|Replicate) + (1|LibraryPrepBatch) + W_1
# No ruv
form_no_ruv <- ~ (1|Individual) + (1|treatment) + (1|Replicate) + (1|LibraryPrepBatch)Partition the variance and plot the result
# Fit model and extract results
# 1) fit linear mixed model on gene expression
# If categorical variables are specified,
# a linear mixed model is used
# If all variables are modeled as fixed effects,
# a linear model is used
# each entry in results is a regression model fit on a single gene
# 2) extract variance fractions from each model fit
# for each gene, returns fraction of variation attributable
# to each variable
# Interpretation: the variance explained by each variables
# after correcting for all other variables
varPart_1_int <- fitExtractVarPartModel( filtered_upperquartile, form, sample_info )Dividing work into 100 chunks...
Total: 29 s# sort variables (i.e. columns) by median fraction
# of variance explained
vp <- sortCols(varPart_1_int)
# violin plot of contribution of each variable to total variance
plotVarPart( vp )
| Version | Author | Date |
|---|---|---|
| e3fbd0f | Anthony Hung | 2021-01-21 |
#noRuv
VarPart_noRUV <- fitExtractVarPartModel( filtered_upperquartile, form_no_ruv, sample_info )Dividing work into 100 chunks...
Total: 28 svp_noRUV <- sortCols(VarPart_noRUV)
plotVarPart( vp_noRUV )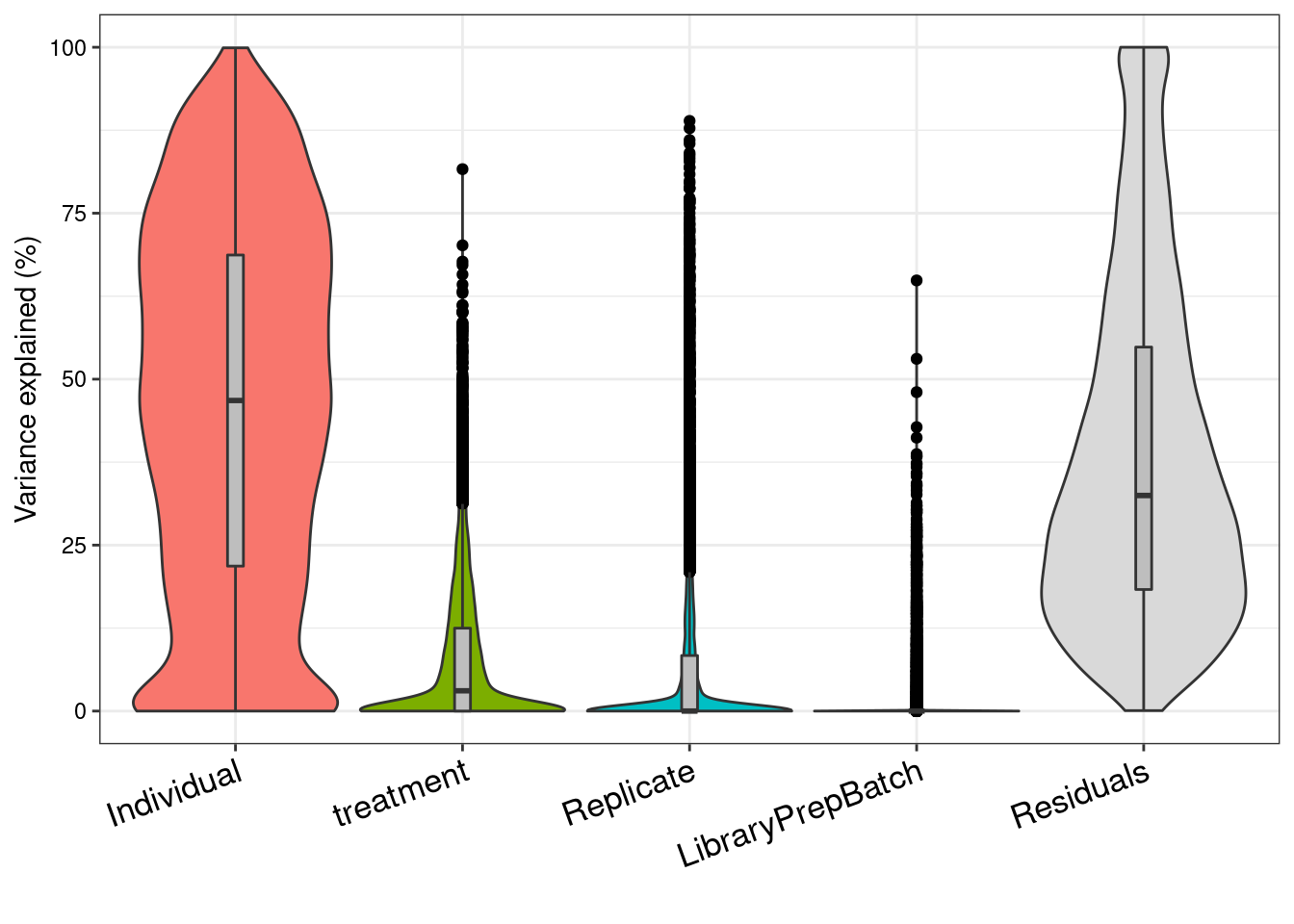
| Version | Author | Date |
|---|---|---|
| e3fbd0f | Anthony Hung | 2021-01-21 |
sessionInfo()R version 3.6.1 (2019-07-05)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Scientific Linux 7.4 (Nitrogen)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.2.19-el7-x86_64/lib/libopenblas_haswellp-r0.2.19.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats4 parallel stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] dplyr_1.0.2 RUVSeq_1.18.0
[3] edgeR_3.26.5 EDASeq_2.18.0
[5] ShortRead_1.42.0 GenomicAlignments_1.20.1
[7] SummarizedExperiment_1.14.1 DelayedArray_0.10.0
[9] matrixStats_0.57.0 Rsamtools_2.0.0
[11] GenomicRanges_1.36.1 GenomeInfoDb_1.20.0
[13] Biostrings_2.52.0 XVector_0.24.0
[15] IRanges_2.18.3 S4Vectors_0.22.1
[17] BiocParallel_1.18.1 variancePartition_1.14.1
[19] Biobase_2.44.0 BiocGenerics_0.30.0
[21] scales_1.1.1 foreach_1.4.4
[23] limma_3.40.6 ggplot2_3.3.3
loaded via a namespace (and not attached):
[1] minqa_1.2.4 colorspace_2.0-0 hwriter_1.3.2
[4] ellipsis_0.3.1 colorRamps_2.3 rprojroot_2.0.2
[7] fs_1.3.1 farver_2.0.3 bit64_0.9-7
[10] AnnotationDbi_1.46.0 codetools_0.2-16 splines_3.6.1
[13] R.methodsS3_1.8.1 doParallel_1.0.14 DESeq_1.36.0
[16] geneplotter_1.62.0 knitr_1.23 workflowr_1.6.2
[19] nloptr_1.2.2.1 pbkrtest_0.4-7 annotate_1.62.0
[22] R.oo_1.24.0 compiler_3.6.1 httr_1.4.2
[25] Matrix_1.2-18 later_1.1.0.1 htmltools_0.5.0
[28] prettyunits_1.1.1 tools_3.6.1 gtable_0.3.0
[31] glue_1.4.2 GenomeInfoDbData_1.2.1 reshape2_1.4.3
[34] Rcpp_1.0.5 vctrs_0.3.6 gdata_2.18.0
[37] nlme_3.1-140 rtracklayer_1.44.0 iterators_1.0.12
[40] xfun_0.8 stringr_1.4.0 lme4_1.1-21
[43] lifecycle_0.2.0 gtools_3.8.1 XML_3.98-1.20
[46] zlibbioc_1.30.0 MASS_7.3-52 aroma.light_3.14.0
[49] hms_0.5.3 promises_1.1.1 RColorBrewer_1.1-2
[52] yaml_2.2.1 memoise_1.1.0 biomaRt_2.40.1
[55] latticeExtra_0.6-28 stringi_1.4.6 RSQLite_2.1.1
[58] genefilter_1.66.0 GenomicFeatures_1.36.3 caTools_1.17.1.2
[61] boot_1.3-23 rlang_0.4.10 pkgconfig_2.0.3
[64] bitops_1.0-6 evaluate_0.14 lattice_0.20-41
[67] purrr_0.3.4 labeling_0.4.2 bit_4.0.4
[70] tidyselect_1.1.0 plyr_1.8.6 magrittr_2.0.1
[73] R6_2.5.0 gplots_3.0.1.1 generics_0.0.2
[76] DBI_1.1.0 pillar_1.4.7 whisker_0.3-2
[79] withr_2.3.0 survival_2.44-1.1 RCurl_1.98-1.1
[82] tibble_3.0.4 crayon_1.3.4 KernSmooth_2.23-15
[85] rmarkdown_1.13 progress_1.2.2 locfit_1.5-9.1
[88] grid_3.6.1 blob_1.2.0 git2r_0.26.1
[91] digest_0.6.27 xtable_1.8-4 httpuv_1.5.1
[94] R.utils_2.9.0 munsell_0.5.0